


JMC | Wang Shaomeng's team: Discovery of a highly potent, orally active ALK inhibitor, CJ-2360, capable of achieving complete tumor regression
Research background
ALK (AnaplasticLymphomaKinase) is a receptor tyrosine kinase belonging to the insulin receptor superfamily.ALK is highly conserved and is expressed predominantly in adult brain tissues, where it plays an important role in the development of the nervous system.In 1994, scientists first identified the mesenchymal large-cell Non-Hodgkin's Lymphoma (ALCL) (NPM)-ALK fusion protein formed by a chromosomal translocation, and subsequently, fusions of ALK with other genes were shown to be present in ALCL, inflammatory myofibroblastoma, diffuse large B-cell lymphoma, squamous cell carcinoma, and non-small cell lung cancer. In addition to chromosomal translocations, ALK gene amplification and point mutation activation of wild-type ALK proteins have also been reported to be present in adult neuroblastomas, ovarian cancers, and inflammatory breast cancers, which makes ALK an attractive target for the treatment of a variety of ALK-containing fusion hematological disorders and solid tumors.
Currently, the following ALK inhibitors are available and in clinical trials: crizotinib (1, crizotinib), ceritinib (2, ceritinib), alectinib (3, alectinib), brigatinib (4, brigatinib), lorlatinib (5, lorlatinib), ASP3026 (6) ( Figure 1).
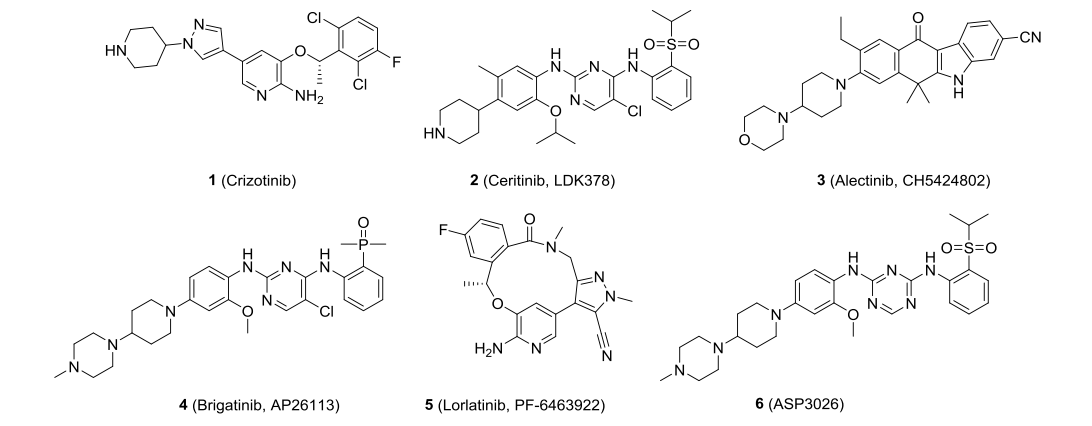
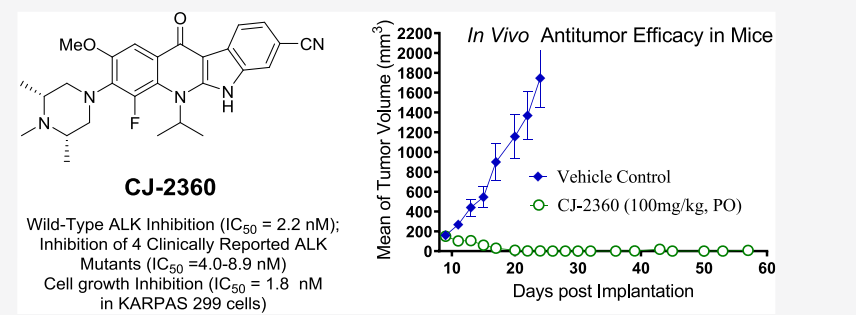
Among them, crizotinib is the first ALK inhibitor approved by the FDA for the clinical treatment of ALK-positive non-small cell lung cancer (NSCLC) patients, but most of the patients are prone to drug-resistant mutations or metastasis leading to deterioration of the disease after 1-2 years of drug administration. Subsequently, ceritinib, alectinib, brigatinib, and lorlatinib have been approved for the treatment of advanced metastatic NSCLC, and ASP3026 is currently in clinical trials.Recently, Wang Shaomeng's team discovered a new highly potent, orally active ALK inhibitor, CJ-2360, which effectively inhibits wild-type ALK kinase and several clinically reported ALK mutants, and achieves complete tumor regression in the ALK-positive KARPAS-299 transplantation tumor model(Figure2).
Experimental content
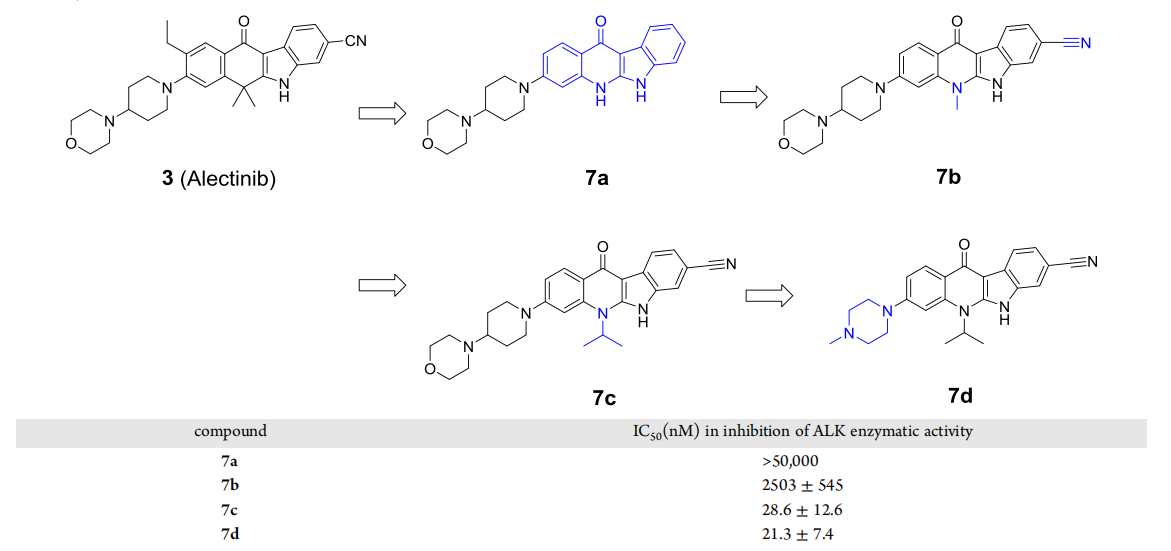
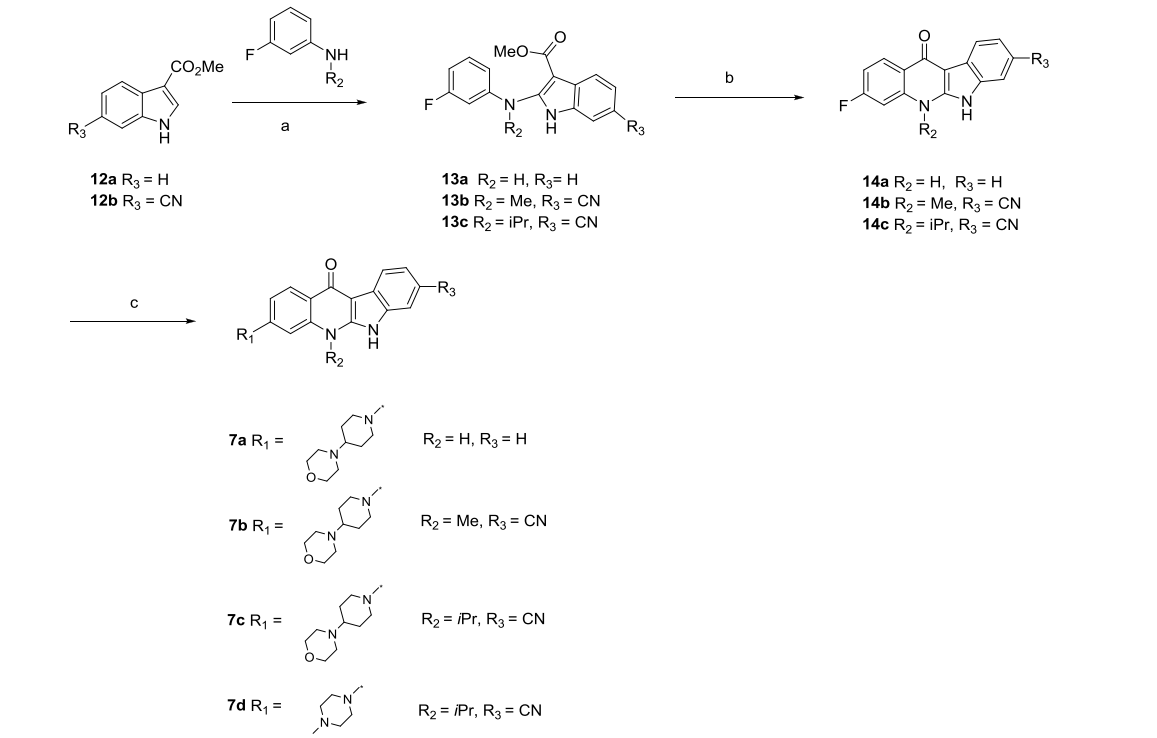
Of all the ALK inhibitors approved for marketing or in the clinical phase, alectinib is the only molecule with a tetracyclic structure, and it has been shown to be selective for ALK over other ALK inhibitors. Therefore, the investigators chose alectinib as a template for designing new ALK inhibitors. In order to preserve the structure of the tetracyclic ring, the researchers designed compounds 7a and 7b using indazoquinoline as the main backbone (Figure 3). In the in vitro enzyme activity assay, 7a showed no inhibitory activity even at a concentration of 50 μM, while 7b showed an IC50 value of 2.5 μM. In the structure of alectinib, there are two methyl groups opposite to the carbonyl group, and the researchers replaced the methyl group at the corresponding position of 7b with an isopropyl group to obtain compound 7c, which reached an IC50 value of 28.6 nM. The major metabolic site of 7c was further was further modified by replacing 4-morpholinopiperidinyl with 1-methylpiperazinyl to obtain compound 7d with better activity (Figure 3).
Referring to the eutectic structure of the complex formed by alectinib and ALK, the researchers modeled the binding of compound 7d with ALK (Figure 4), which has a similar binding pattern, with the more significant difference that one ethyl group on the tetracyclic concatenated backbone of alectinib has hydrophobic interactions with residues L1198, A1200, and L1122 on ALK, and the compound 7d does not have any substituent at the same position.
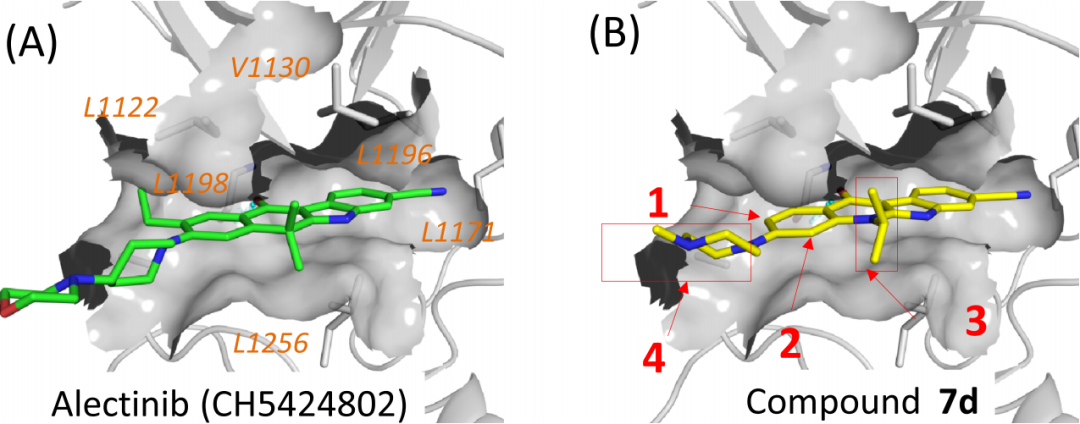
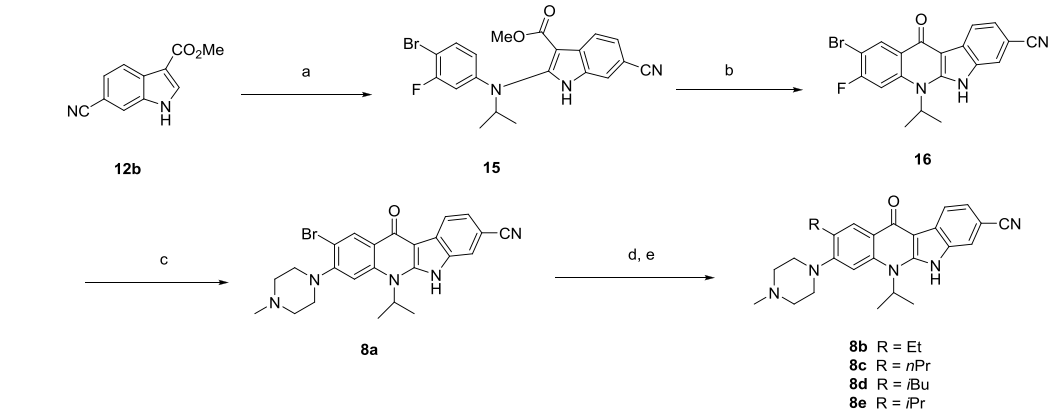
As shown by the arrows in Figure 4-B, introduction of an ethyl group at position 1 of compound 7d yielded compound 8b, which showed a 10-fold enhancement in inhibitory activity compared to 7d (Figure 5-8bvs7d), and all alkyl substitutions or bromine substitutions except isopropyl substitutions gave good IC50 values (Figure 5-8a-8e). The researchers then substituted alkyl substitutions with alkoxy substitutions to synthesize compounds 8f-8i, all of which showed better inhibition of ALK than 7d. What's more, the researchers evaluated the inhibitory activity of the above compounds on cell proliferation in the KARPAS-299 cell line harboring ALK fusion proteins, and all of them showed a significant increase in their effects compared to 7d.
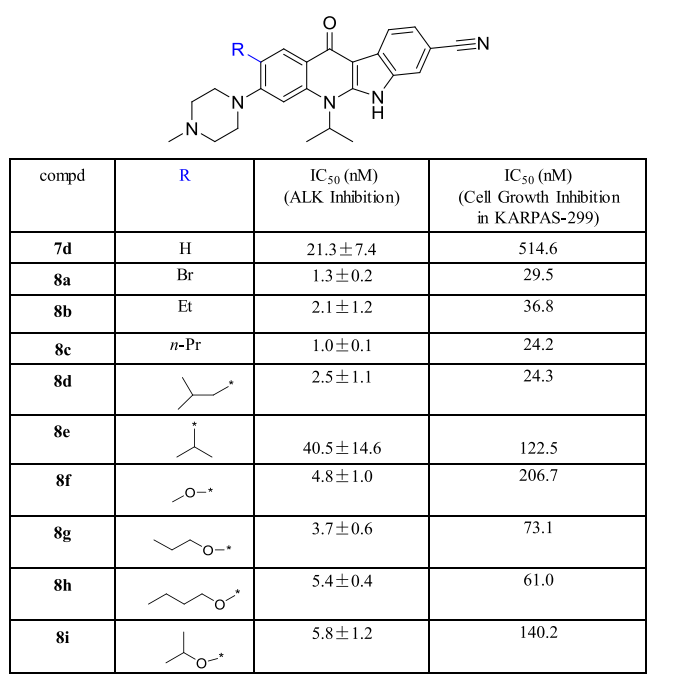
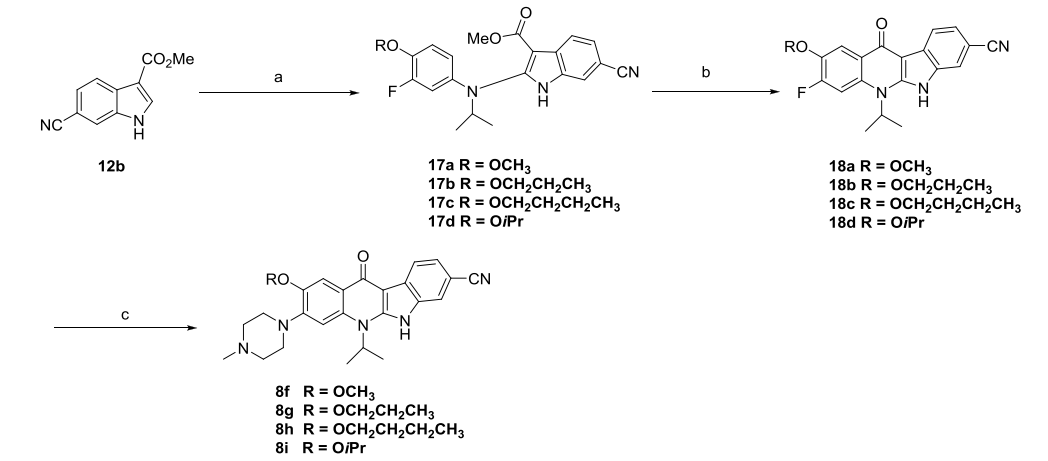
As shown by the arrows in Figure 4-B, the introduction of fluorine atom at position 2 of compound 7d yielded compound 9a, whose inhibitory ability against ALK was comparable to that of 7d, but 9a showed a 10-fold increase in activity compared to 7d in the KARPAS-299 cell line (Figure 6). Using 8f as a template compound and introducing a fluorine atom at its position 2 to obtain compound 9c also showed a 10-fold increase in cellular activity compared to 8f. These data indicate that the introduction of fluorine atoms on the benzene ring can significantly increase the cellular potency of the compounds.
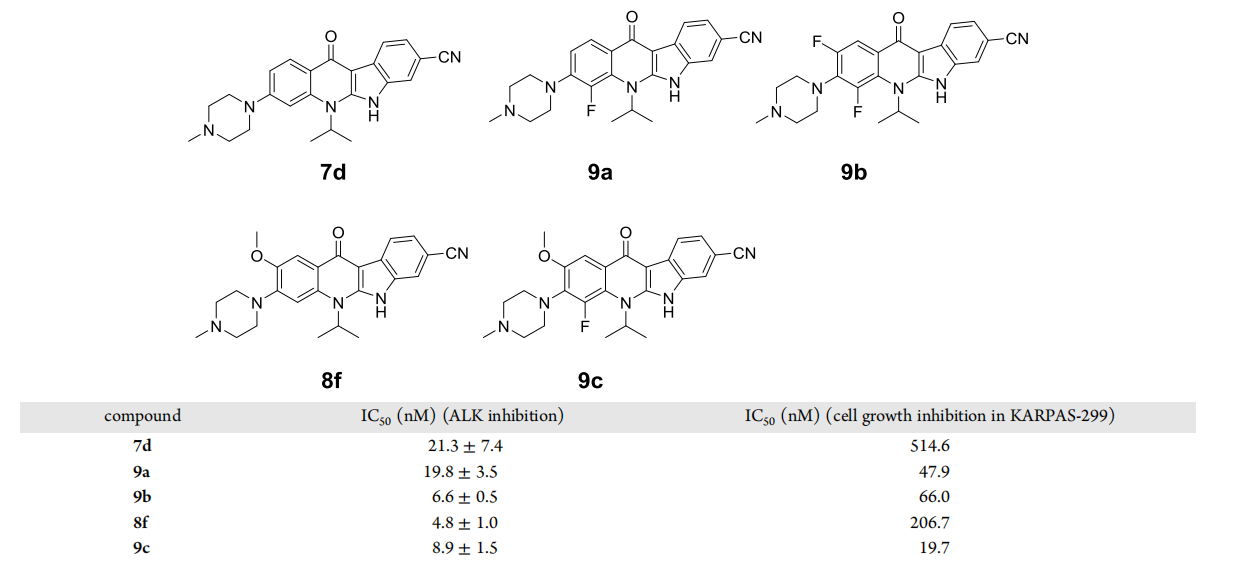
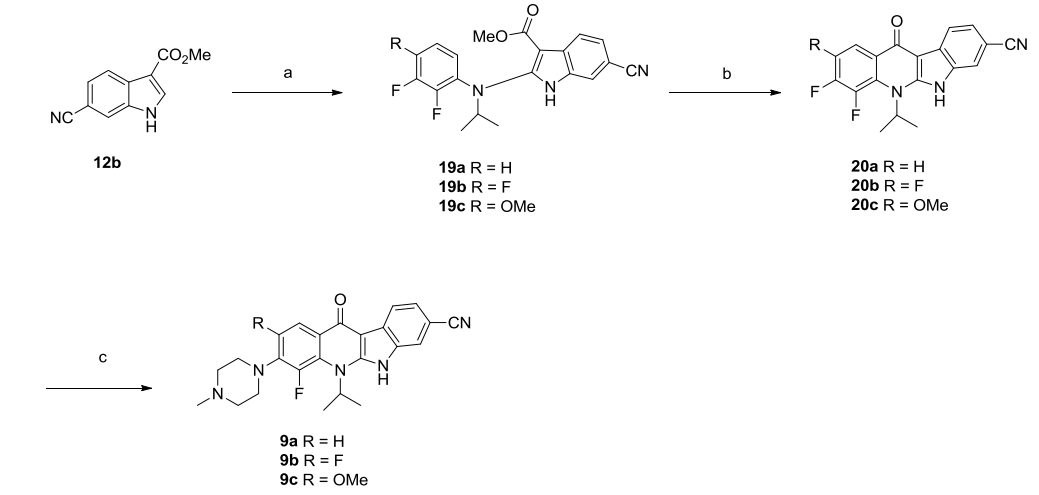
The substituents on the quinoline nitrogen were examined using compound 8f as a template (Figure 7), replacing isopropyl with other aliphatic chains such as ethyl, propyl, sec-butyl, or 2-pentyl yielded compounds 10a-10d, all of which showed varying degrees of decreased inhibitory activity against ALK compared to 8f; when isopropyl was replaced with cycloalkyls, particularly cyclobutyl, cyclohexyl or cyclohexyl, they exhibited very similar cell proliferation inhibitory activities in the KARPAS-299 cell line exhibited very similar cell proliferation inhibitory activities.
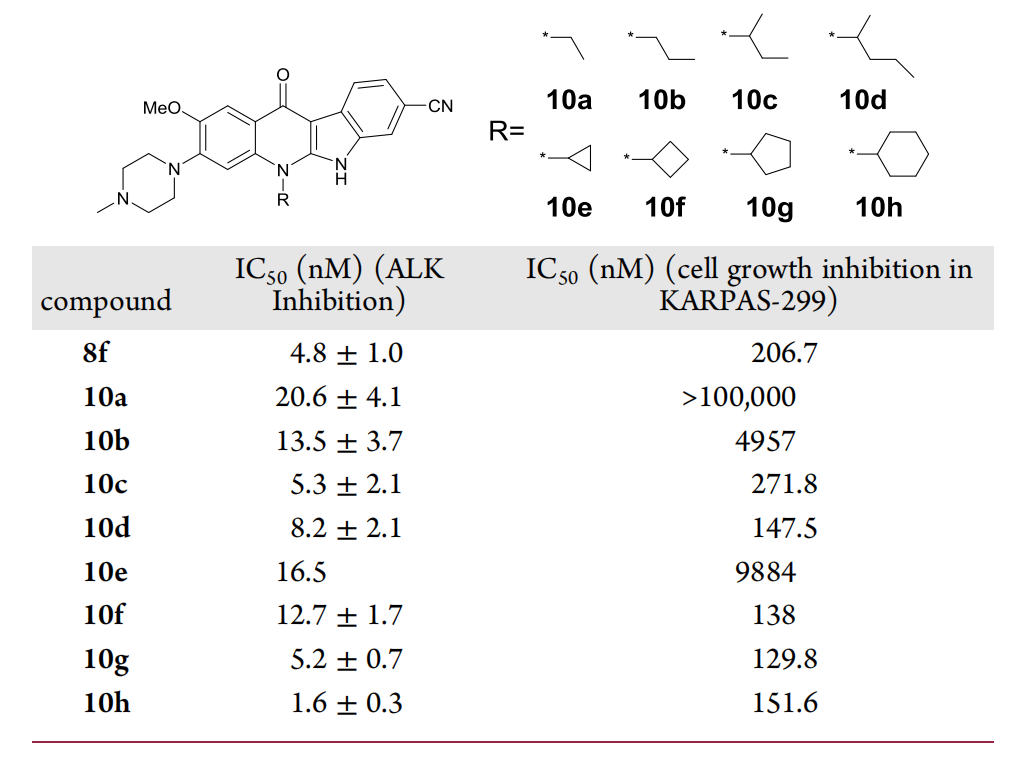
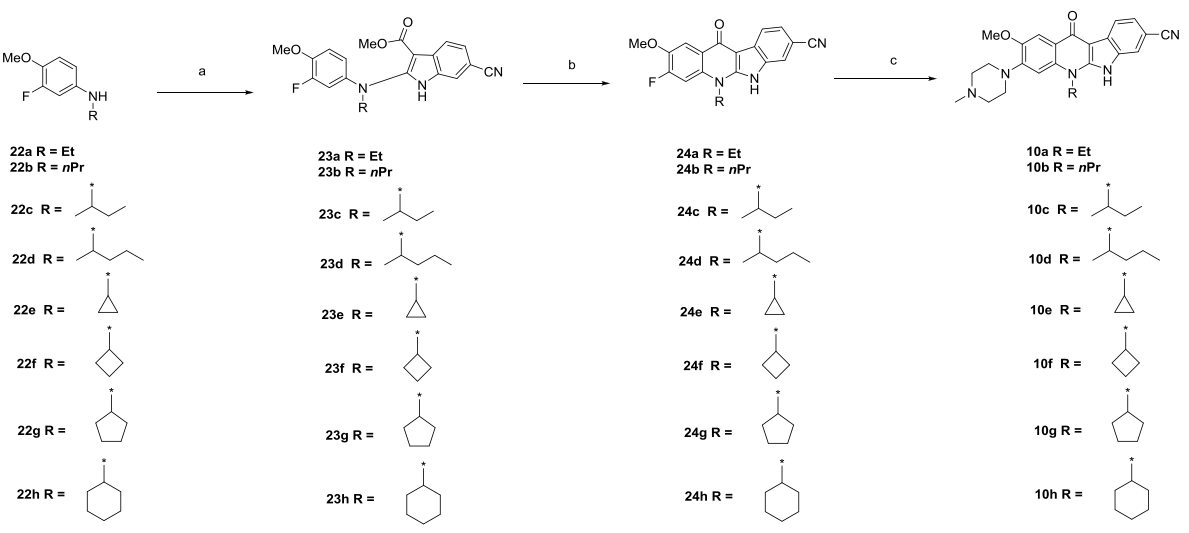
Finally, using 9c as a template, the researchers examined the effect of different solubilizing groups at position 4 on the activity of the molecule (Fig. 8), and finally obtained the best active compound, CJ-2360, which not only exhibited strong ALK inhibition, but also reached an IC50 value of 1.8 nM for inhibiting cell proliferation in the KARPAS-299 cell line.
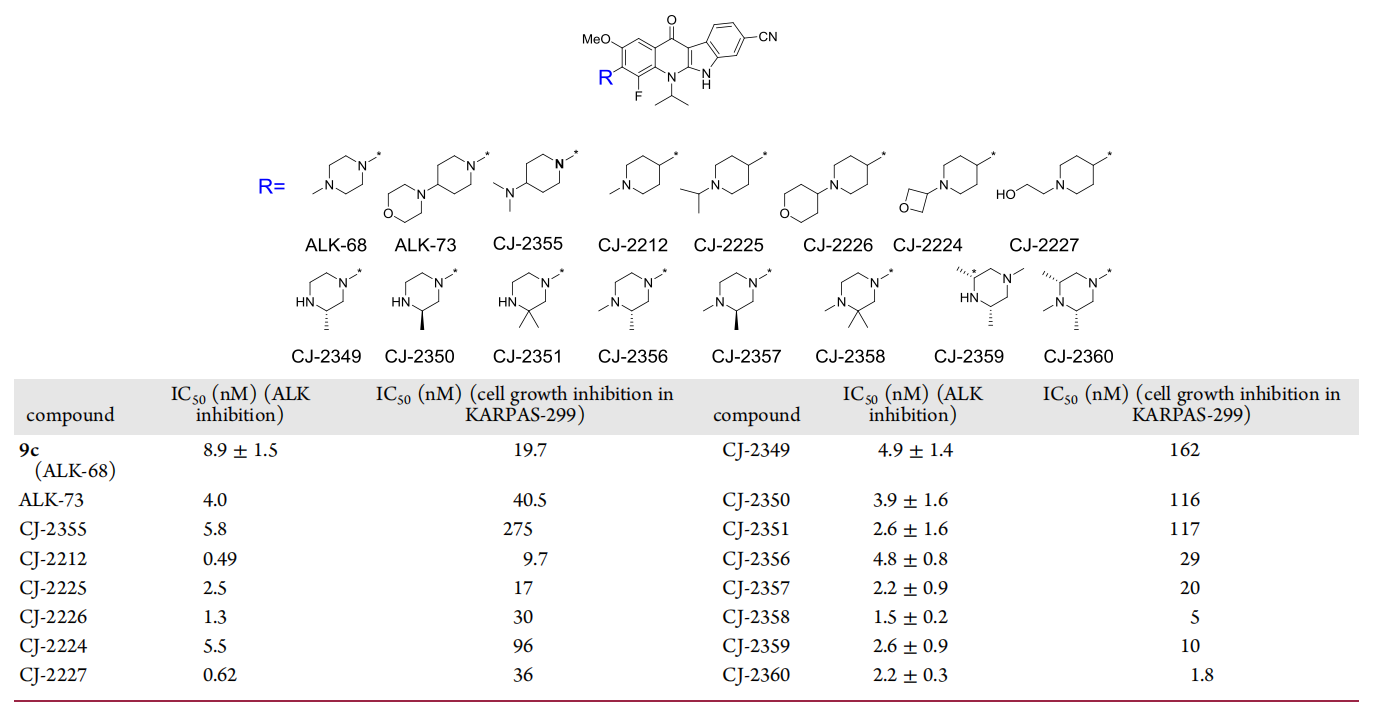
Among the many compounds examined, CJ-2360 exhibited the best combined enzymatic and cellular activities, and subsequently, the investigators evaluated the pharmacokinetic parameters of CJ-2360 in mice and rats, respectively (Figure 9). The results showed that after oral administration, CJ-2360 achieved a reasonable oral exposure in mice, with an overall oral bioavailability of 38.2%, a moderate plasma clearance (1.2 L/h/kg), and a large volumetric distribution (7.5 L/kg) suggesting a wide tissue distribution of CJ-2360 in mice. In vivo pharmacokinetic studies in rats gave similar data.
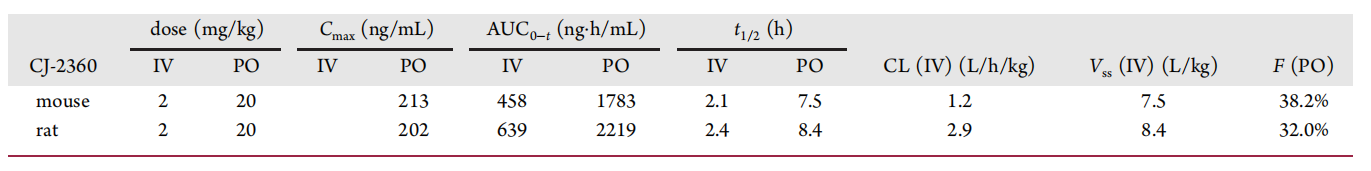
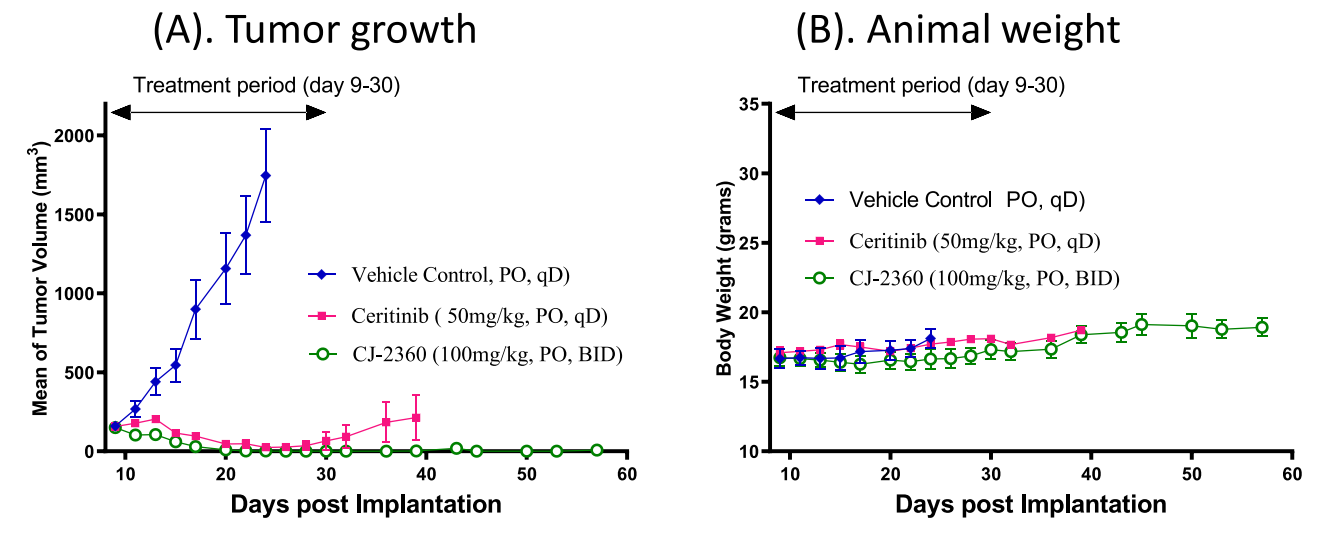
In addition, the investigators administered CJ-2360 orally to mice bearing KARPAS-299 transplanted tumors and analyzed the drug concentration of CJ-2360 in plasma and tumor tissues of the mice, confirming that good plasma exposure of CJ-2360 was obtained after a single oral administration in mice, and that its concentration in tumor tissues was much greater than that in plasma.Based on the good in vivo pharmacokinetic data and tumor tissue exposure of CJ-2360, the investigators evaluated its antitumor effects in mice bearing KARPAS-299 transplant tumors and used ceritinib as a positive control (Figure 10). The data showed that CJ-2360 was able to achieve complete regression of all tumors after twice-daily administration, and the tumors did not start to recover until the 23rd day after the last administration, demonstrating that CJ-2360 was able to achieve complete and long-lasting tumor regression in mice bearing KARPAS-299 transplanted tumors; moreover, the mice tolerated CJ-2360 well, and did not induce weight loss or other signs of toxicity throughout the experiment. In contrast, ceritinib achieved only partial tumor regression.
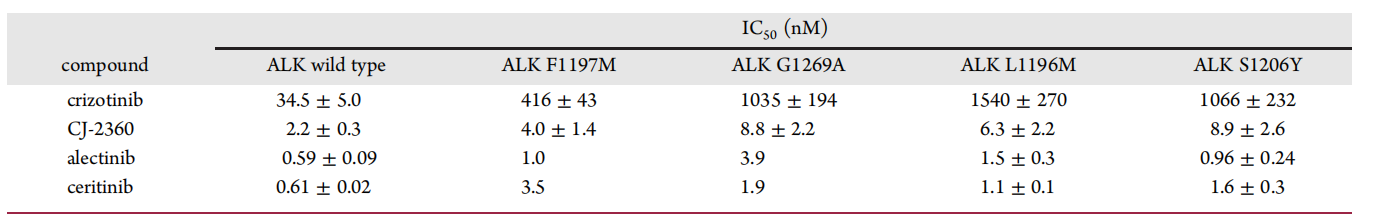
In the clinic, some patients with ALK-positive tumors develop resistance due to ALK mutations after treatment with crizotinib, and several clinical ALK mutants have been identified. During the development of ALK inhibitors, their ability to inhibit various ALK mutants must be examined; therefore, the investigators compared the inhibitory activities of several marketed ALK inhibitors and CJ-2360 against ALK mutants (Figure 11). The results showed that CJ-2360 also had high inhibitory activity against ALK mutants including F1197M, G1269A, L1196M, and S1206Y, reaching 4.0, 8.8, 6.3, and 8.9 nM, respectively.
Summarize
Using alectinib as a template compound, the researchers designed and synthesized a series of novel ALK inhibitors containing the indazoquinoline backbone, and evaluated them to select the compound with the best combination of enzyme and cellular activity, CJ-2360, which has good oral plasma exposure, tumor tissue concentration, and bioavailability, and has high inhibitory activity against wild-type ALK and several clinical ALK mutants. mutants with high inhibitory activity. Most importantly, in vivo antitumor assays demonstrated that CJ-2360 achieved complete and durable regression of all ALK-positive tumors, both wild-type and mutant, which was superior to that of ceritinib, suggesting that CJ-2360 is a promising novel ALK inhibitor for late-stage preclinical studies.
Compound synthesis
1. Synthesis of compounds 7a-7d
2. Synthesis of compounds 8a-8e
3. Synthetic compounds 8f-8i
4. Synthesis of compounds 9a-9c
5. Synthesis of compounds 10a-10h
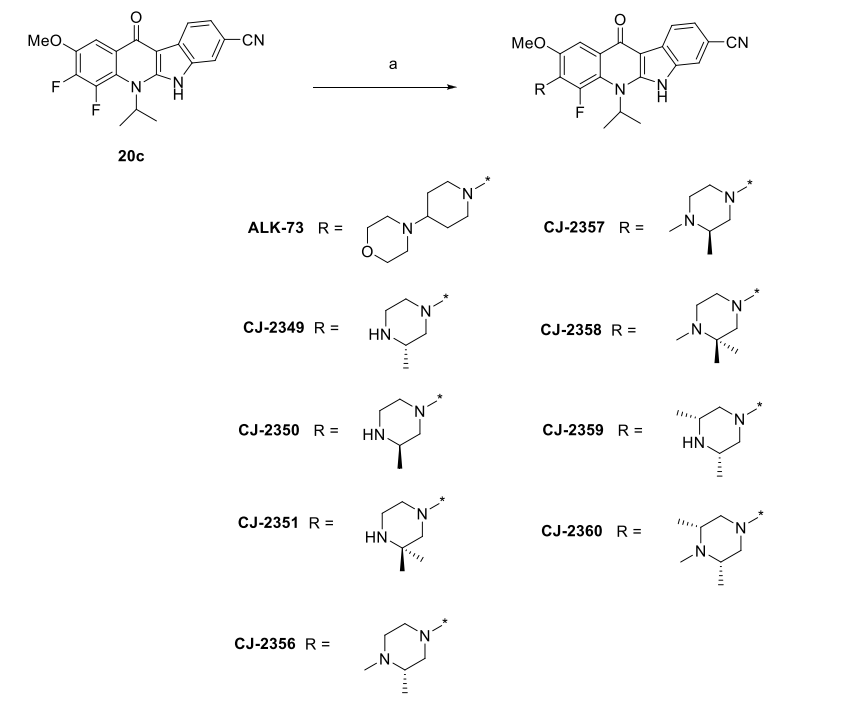
6. Synthetic ALK-73, CJ-2349-2351 and CJ-2356-2360
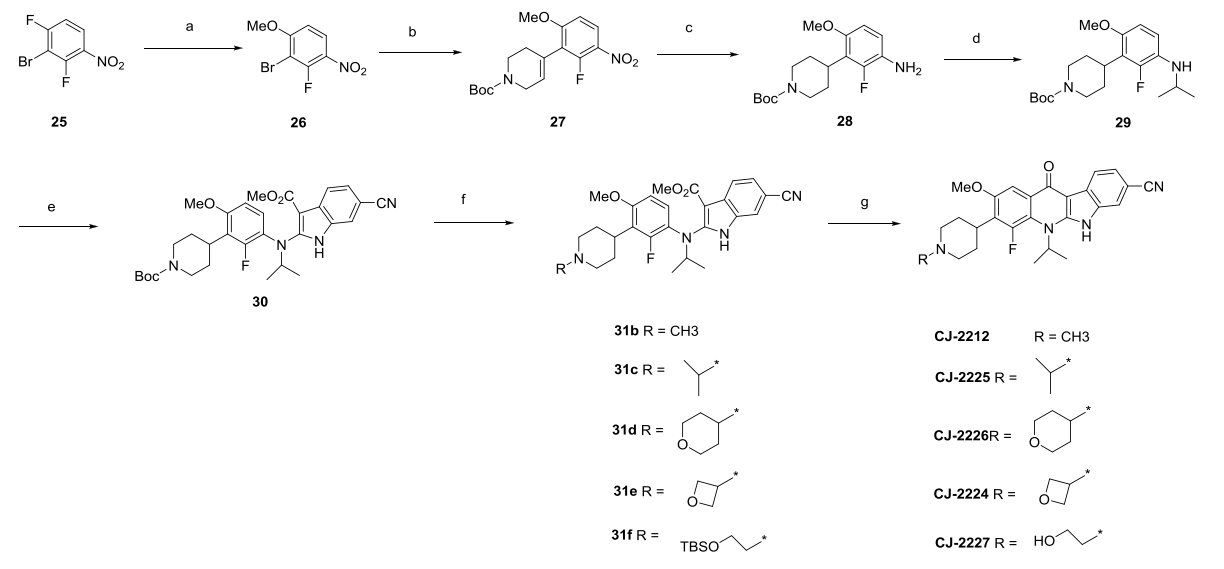
7. Synthesis of CJ-2212 and CJ-2224-2227
References
JianyongChen, Yunlong Zhou, Xuyuan Dong, Liu Liu, Longchuan Bai, DonnaMcEachern, Sally Przybranowski, Chao-Yie Yang, Jeanne Stuckey,Xiaoqin Li, Bo Wen, Ting Zhao, Siwei Sun, Duxin Sun, Lingling Jiao,Yu Jing, Ming Guo, Dajun Yang, and Shaomeng Wang. Discovery of CJ-2360 as a Potent and Orally Active Inhibitor of Anaplastic Lymphoma Kinase Capable of Achieving Complete Tumor Regression. Journal of Medicinal Chemistry (2020),63(22),13994-14016.Note: Images in the text are from references
Recommended Products
| CS-D1229 | CS-0045068 | CS-0140671 | |||
 |
 |
 |
|||
| CS-W008832 | CS-0054334 | CS-0127903 | |||
 |
 |
 |
|||
| CS-B0412 | CS-0140673 | CS-W004420 | |||
 |
 |
 |